In our group we investigate chemical problems related to the structure, spectroscopy, dynamics and function of molecular systems. Our area of research combines ab initio electronic structure theory and Density Functional Theory with reaction dynamics. The marriage of these fields allows us to understand chemical processes, not only from a stationary point of view, but also dynamically. Our expertise focuses on the accurate treatment of molecular systems after light irradiation but we also study interesting chemical reactions that occurs in the electronic ground state.
Computational photochemistry and spectroscopy
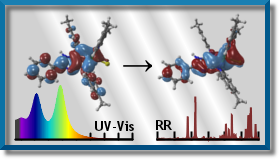
We use computational chemistry to predict IR, Resonance Raman (RR), and UV-vis spectra. We aim at designing photonic materials with applications in photobiology, photomedicine and solar energy conversion devices.

Femtosecond spectroscopy and molecular movies
The goal is simulating intra- and inter-molecular dynamics of light-induced processes on the shortest time-scales. We characterize potential energy surfaces to understand chemical reactivity and relaxation mechanisms at molecular level.
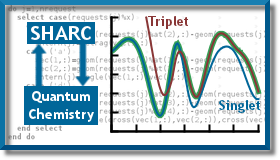
Development of quantum/classical methods to describe general ultrafast photoinduced dynamical processes
Our main interest here is to develop a general code (SHARC) which can model all kind of photochemical processes, involving singlet and triplet states, as well as any type of coupling.
Simulation of drugs in biological environments
The goal is to achieve fundamental understanding of the mode of action of drugs interacting with biological residues by means of force field and ab initio molecular dynamics simulations. The therapeutic mechanisms may occur in the electronic ground state or involve several excited states.
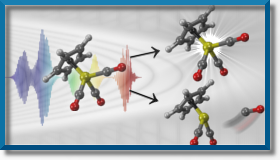
Laser control of chemical reactions
The aim is to obtain laser pulses which can induce a desired chemical reaction. Examples include selective bond breaking, isomerization, hydrogen/proton transfer, ionization, as well as controlling molecular switches and driving molecular machines.